


롱 레이지 상호작용에 대한 포괄적 매핑이 인간 유전체 접힘의 원리를 밝히다
(세포핵내 유전체 접힘의 3차원 구조)

- 우리는
근접성-기반 접합 (proximity-based ligation)과
대량 병렬 시퀀싱 (massively parallel sequencing)을 결합하여,
전체 게놈의 3차원 구조 (three-dimensional architecture of whole genomes)를 조사하는 방법인
Hi-C를 설명한다. - 우리는 1 megabase의 해상도에서 Hi-C로 인간 게놈의 공간 근접성 맵 (spatial proximity maps)을 구축했다.
- 이 맵은
염색체 영역의 존재 (presence of chromosome territories)와
작고 유전자가 풍부한 염색체(small, gene-rich chromosomes)의 공간 근접성(spatial proximity)을
확인한다. - 우리는
두 개의 게놈 전체 구획(two genome-wide compartments)을 형성하기 위해,
오픈 크로마틴(open chromatin)과 닫힌 크로마틴(closed chromatin)의 공간적 분리(spatial segregation)를
특징으로 하는 추가 수준의 게놈 조직을 식별했다. - 메가베이스 규모에서 크로마틴 형태(chromatin conformation)는 프랙탈 구형체(fractal globule)와 일치한다.
- 프랙탈 구형체는
매듭이 없는(knot-free) 폴리머 형태(polymer conformation)로,
모든 게놈 위치(locus)를 쉽게 접고 펼칠 수 있는 능력을 유지하면서도
최대한 밀집된 패킹을 가능하게 한다. - 프랙탈 구형체는 보다 일반적으로 사용되는 globular equilibrium model과 다르다.
- 우리의 결과는 전체 게놈의 동적 형태(dynamic conformations)를 매핑하는 Hi-C의 힘을 보여준다.

- 염색체의 3차원(3D) 형태(conformation)는
핵을 구획화(compartmentalizing)하고, 멀리 떨어진 기능적 요소들을
가까운 공간적 근접성(close spatial proximity)으로 가져오는 데 관여한다 [1–5]. - 염색체가 접히는 방식을 이해하면,
크로마틴 구조 (chromatin structure),
유전자 활동 (gene activity) 및
세포의 기능 상태 (functional state of the cell) 간의 복잡한 관계에 대한 통찰력을 얻을 수 있다. - 그러나 핵소체 (nucleosomes)의 규모를 넘어서 크로마틴 조직에 대해서는 알려진 바가 거의 없다.
- 특정 쌍의 유전자좌(pairs of loci) 간의 장거리 상호작용(Long-range interactions)은
공간적으로 제한된 접합(spatially constrained ligation)을 사용한 다음,
유전자좌-특유적 (locus-specific) 중합효소 연쇄 반응 (polymerase chain reaction)(PCR)을 사용하여,
염색체 구조 포착 (chromosome conformation capture, 3C)으로 평가할 수 있다 [6]. - 3C의 조정(adaptation)은
inverse PCR (4C) [7, 8] 또는
다중 접합-매개 증폭(multiplexed ligation-mediated amplification, 5C) [9]을 사용하여
프로세스를 확장했다. - 그래도 이러한 기술은 일련의 대상 유전자좌(target loci)를 선택해야 하며
不偏的 (unbiased) 게놈-전체 분석을 허용하지 않는다. - 여기서 우리는, Hi-C라는 방법을 보고하는 바, 이는 위의 어프로치를 조정하여
접합 생성물(ligation products)을 정제(purification)한 다음, 대량 병렬 시퀀싱 (massively parallel sequencing)을 가능하게 한다. - Hi-C는 전체 게놈에서 크로마틴 상호작용을 편향 없이 식별할 수 있게 한다. 우리는 이 과정을 간략하게 요약한다 :
🧬 세포들은 포름알데히드로 교차 고정된다 (crosslinked);
🧬 DNA는 5′ overhang (돌출부)을 남기는 제한 효소로 분해된다;
🧬 5' 오버행은 바이오틴화된 잔류물(biotinylated residue)을 포함하여 채워진다;
🧬 그리고 그 결과 나온 뭉툭한-말단 단편들(blunt-end fragments)은
교차 고정된 DNA 단편들 사이의 접합 이벤트(ligation events)를 선호하는
희석된 조건(dilute conditions)에서 접합된다.
🧬 그 결과 나온 DNA 샘플에는 핵에서 원래 공간적으로 가까운 곳에 있었던 단편들로 구성된
접합 생성물(ligation products)이 포함되어 있으며, 접합부에서 바이오틴으로 표시된다.
🧬 Hi-C 라이브러리는 DNA를 전단(shearing)하고
스트렙타비딘 비드(streptavidin beads)로 바이오틴이 함유된 단편들을 선택하여 생성된다.
🧬 그런 다음 그 라이브러리는 대량 병렬 DNA 시퀀싱(massively parallel DNA sequencing)을 사용하여 분석되어,
상호작용하는 단편들의 카탈로그를 생성한다 (Fig. 1A) [10].

Fig. 1. Overview of Hi-C.
(A) 세포들이 포름알데히드로 크로스-링크되어, 공간적으로 인접한 크로마틴 세그먼트들 간에 공유 연결(covalent links)이 된다. (DNA 단편들이 진청색, 빨간색으로 보인다; 그런 상호작용을 가능하게 할 수 있는 단백질들은 연청색과 청록색으로 보인다). 염색질(chromatin)이 제한효소(restriction enzyme)로 분해되고 (여기서는 HindIII; 점선으로 표시된 제한 부위; 삽도를 보라), 그 결과 달라붙는 말단들(sticky ends)이 뉴클레오티드들에 채워지고 그 중 하나는 비오틴화된다 (핑크색 점). 접합(Ligation)이 극히 희석된 조건 하에서 수행되어, 키메라 분자들(chimeric molecules)을 생성한다; HindIII 부위가 손실되고 NheI 부위가 생성된다 (삽도). DNA가 정제되고 전단된다. 비오틴화된 접합부는 스트렙타비딘 비드 (streptavidin beads)로 분리되고 페어드 엔드 시퀀싱(paired-end sequencing)으로 식별된다.
(B) Hi-C는 genome-wide contact matrix를 생성한다. 여기서 보여지는 서브 매트릭스는 염색체 14번에서의 염색체 내 상호작용들에 해당한다 (염색체 14번은 acrocentric하다(말단동원체, 동원체가 염색체 완이 다른 것들보다 훨씬 더 짧게 위치함; 그 짧은 완은 보이지 않는다). 각 픽셀은 1-Mb locus와 다른 1-Mb locus 간의 모든 상호작용들을 나타낸다; intensity는 reads의 총수이다 (0에서 50). 체크 마크들은 10 Mb마다 나타난다.
(C and D) 우리는 오리지날 실험을 동일한 제한효소를 사용한 생물학적 repeat로부터의 결과 [(C), 0에서 50 reads까지)]와, 다른 제한효소를 사용한 결과 [(D), Ncol, 0에서 100 reads까지]와 비교했다.
- 우리는 핵형적으로 정상적인 인간 림프모세포주(lymphoblastoid cell line) (GM06990)에서
🧬 Hi-C 라이브러리를 만들고,
🧬 Illumina Genome Analyzer (Illumina, San Diego, CA)의 두 레인에서 시퀀싱하여,
🧬 인간 게놈 레퍼런스 시퀀스에 고유하게 정렬될 수 있었던 840만 개의 리드 쌍을 생성했다. - 이 중 670만 개는
20kb 이상 떨어진 세그먼트들 간의 장거리 접촉(long-range contacts)에 해당했다. - 우리는 게놈을
🚀 1-Mb 영역들("loci")로 나누고,
🚀 매트릭스 엔트리 mij를 locus i와 locus j 사이의 접합 생성물(ligation products)의 수로 정의하여
🚀 게놈 전체 접촉 매트릭스 (genome-wide contact matrix) M을 구축했다 [10]. - 이 매트릭스는
➡ 원래 세포 샘플에 존재하는 상호작용의 앙상블 평균을 반영한다;
➡ 시각적으로 히트맵(heatmap)으로 표현될 수 있으며,
➡ intensity는 접촉 빈도 (contact frequency)를 나타낸다 (Fig. 1B). - 우리는 같은 제한효소 (HindIII)로 그리고 다른 제한효소 (NcoI)로 실험을 반복하여
Hi-C 결과가 재현될 수 있는지 테스트했다. - 우리는 이들 새로운 라이브러리들 (Fig. 1, C and D)의 contact 매트릭스들이 오리지널 contact 매트릭스와 매우 비슷했다는 것을 관찰했다 [Pearson’s ρ = 0.990 (HindIII) and ρ = 0.814 (NcoI); P was negligible (<10–300) in both cases].
- 그러므로 우리는 이 세가지 데이터 세트를 후속 분석들에서 결합하였다.
- 우리는 먼저 우리의 데이터가 알려진 게놈 조직 특징[1]과 일치하는지 여부를 테스트했다:
구체적으로는
🧬 염색체 영토들 (chromosome territories) (동일한 염색체 상의 먼 loci가 공간에서 서로 가까이 있는 경향)과
🧬 핵내 위치 패턴 (특정 염색체 쌍이 서로 가까이 있는 경향)이다. - 우리는 염색체 n에서
게놈 거리 s (뉴클레오티드 시퀀스를 따라 염기쌍으로 표시된 거리)로 분리된
유전자좌 쌍 (pairs of loci)에 대한
평균 염색체 내 접촉 확률 (average intrachromosomal contact probability) In(s)를 계산했다. - In(s)는 모든 염색체들에서 단조적으로 감소하여, 유전자좌 사이의 3D 거리가 게놈 거리(genomic distance) 증가에 따라 증가하는 폴리머와 같은 행태를 시사한다; 이러한 결과는 3C 및 fluorescence in situ hybridization (FISH)과 일치한다 [6, 11].
- 200Mb 보다 더 큰 거리들에서도, In(s)는 서로 다른 염색체들 간의 평균 접촉 확률보다 항상 훨씬 더 크다 (Fig. 2A).
이는 염색체 영토들의 존재 (existence of chromosome territories)를 의미한다.

Fig. 2 염색체 영토의 존재와 조직.
(A) 접촉 확률은 염색체 1의 게놈 거리에 따라 감소하여 결국 ~90Mb(파란색)에서 정점에 도달한다. 염색체 간 접촉 수준(검은색 대시)은 염색체 쌍마다 다르다. 염색체 1의 유전자좌는 염색체 10의 유전자좌(녹색 대시)와 상호작용할 가능성이 가장 높고, 염색체 21의 유전자좌(빨간색 점선)와 상호작용할 가능성이 가장 낮다. 염색체 간 상호 작용은 염색체 내 상호 작용에 비해 감소한다.
(B) 모든 염색체 쌍들 사이의 관찰/예상 염색체 간 접촉 수. 빨간색은 풍부함을 나타내고 파란색은 감소를 나타낸다 (범위 0.5~2). 작고 유전자가 풍부한 염색체는 서로 더 많이 상호작용하는 경향이 있어 핵에서 함께 클러스터링된다는 것을 시사한다.
- 염색체 쌍들 간의 염색체간 접촉 확률 (Interchromosomal contact probabilities) (Fig. 2B)은
작고, 유전자가 풍부한 염색체들 (염색체 16, 17, 19, 20, 21, and 22)이
서로 더 잘 상호작용한다는 것을 보여준다. - 이것은 이런 염색체들은 핵의 중심부에 자주 서식한다는 것을 보여주는 FISH 연구들과 일치한다 [12, 13].
- 흥미롭게도 작지만 유전자가 많지 않은 염색체 18은 다른 작은 염색체들과 자주 상호작용하지 않는다;
이는 염색체 18은 핵의 주변부 근처에 위치하는 경향이 있다는 것을 보여주는 FISH 연구들과 일치한다 [14]. - 그런 다음 우리는 서로 잘 연관되는 염색체 영역들(chromosomal regions)이 있는지 여부를 탐구하기 위해
개별 염색체들에 대해 확대해보았다. - 시퀀스 근접성이 접촉 확률에 강하게 영향을 미치기 때문에, 우리는
접촉 매트릭스 내의 각 엔트리를,
그 게놈 거리에 있는 유전자좌들의 게놈-전체 평균 접촉 확률(genome-wide average contact probability)로
나누어서
정규화된 접촉 매트릭스(normalized contact matrix) M*를 정의했다 [10].
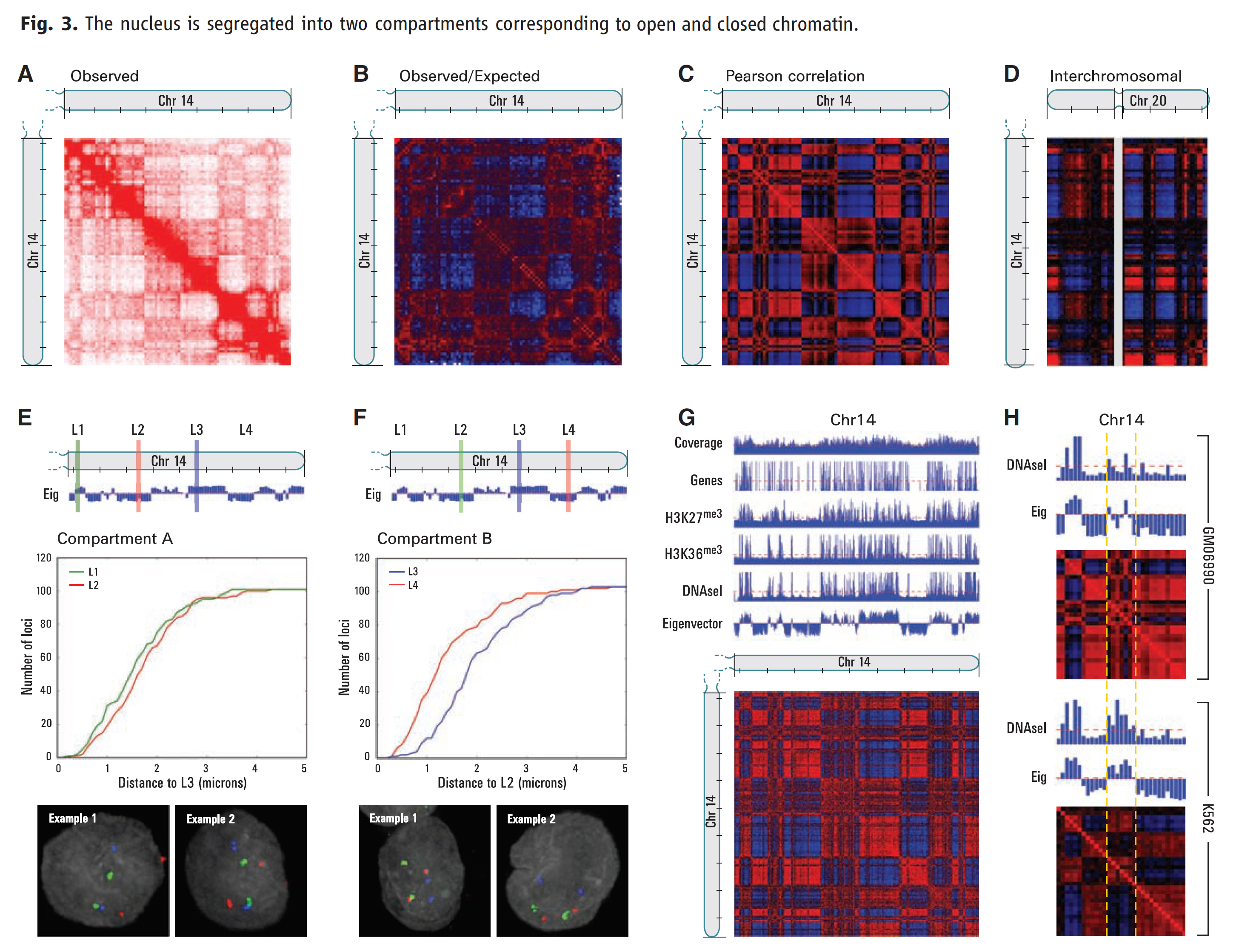
Fig. 3. 핵(nucleus)은 open and closed chromatin에 해당하는 2개의 구획들(two compartments)로 구분된다.
(A) 염색체 14, 1 Mb 해상도의 맵이 강렬한 대각선과 큰 블록들의 무리의 형태로 하위구조를 보임 (3개 실험들이 결합; 0에서 200 reads까지 범위). 체크 마크는 10 Mb마다 나타남.
(B) observed/expected matrix가 주어진 게놈 거리에서 (0.2에서 5까지 범위) 기대되는 것 보다 더 많거나 (빨간색) 적은(청색) 상호작용들을 가진 유전자좌들을 보여줌.
(C) 염색체 14를 따라 1-Mb 유전자좌 쌍들의 염색체내 상호작용 프로파일들 간의 상관관계를 보여줌 [– (blue)에서 +1 (red)까지의 범위]. 격자무의 패턴은 그 염색체 내에 2개 구획들의 존재를 가리킴.
(D) 염색체 20 주위의 접합불가 영역이 회색으로 표시됨. 염색체 14에서의 각 구획은 매우 비슷한 게놈 전체 상호작용 패턴을 가진 염색체 20에서의 카운터파트가 있음.
(E and F) 우리는 염색체 14를 따라 연속적으로 있지만 두 구획들 사이에서 번갈아 나타나는 4개의 유전자좌 (L1, L2, L3, and L4)에 대한 조사를 설계했음 [구획A의 L1과 L3; 구획B의 L2와 L4]. (E) L2가 그 게놈의 주요 시퀀스에서 L1과 L3 사이에 위치한다는 사실에도 불구하고, L3 (청색)가 L2 (적색)보다 L1 (녹색)에 일관되게 더 가까웠음. 이는 시각적으로 그리고 누적 분포 플롯팅에 의해서 확인되었음. (F) L2 (녹색)가 L3(청색)보다 L4 (적색)에 더 가까웠음.
(G) 염색체 14, 100 kb 해상도의 상관관계 맵. PC (eigenvector)는 유전자 분포와 오픈 크로마틴의 특징들과 상관됨.
(H) 염색체 14로부터의 31-Mb 윈도우가 보임; 표시된 영역 (황색 점선)은 GM06990에서 오픈 및 크로즈드 구획들 사이에서 번갈아 나타나지만 (top, eigenvector and heatmap), K562에서는 주로 오픈에서 나타남 (bottom, eigenvector and heatmap). 구획화에서의 변화는 크로마틴 상태에서의 이동에 해당됨 (DNAseI).
- 정규화된 매트릭스는
풍부한 상호작용들 및 감손된 상호작용들(enriched and depleted interactions)의 많은 대형 블록들을 보여주며,
이를 통해 격자무늬 패턴 (plaid pattern)을 생성한다 (Fig. 3B). - 만일 2개의 유전자좌들 (여기서는 1-Mb regions)이 공간에서 인근에 있으면,
우리는 그들은 이웃들을 공유할 것이며 상관적인 상호작용 프로파일들을 가질 것이라고 추론했다. - 그러므로 우리는 상관계수 매트릭스C를
cij가 M*의 i번째 행과 j번째 열 사이의 Pearson correlation인 것으로 정의했다. - 이 과정은 격자 무늬 패턴을 극적으로 선명하게 만들었다 (Fig. 3C);
결과로 나온 매트릭스 엔트리들의 71%가 통계적으로 유의한 상관계수들을 나타낸다 (P ≤ 0.05). - 이 격자 무늬 패턴은,
각 염색체가 두 유전자좌들의 세트들로 나뉘어 질 수 있다는 것을 제시하는데 (임의로 A와 B라고 표시),
각 세트 내에 접촉들은 풍부하고, 세트들 간의 접촉들은 대폭 감소되도록 나누어 질 수 있다. - 우리는 principal component analysis를 사용하여 이런 식으로 각 염색체를 분할했다.
- 두개의 염색체를 제외한 모든 염색체에서 첫 번째 주성분(PC)은
격자 무늬 패턴과 명확히 일치했다 (양의 값들이 한 세트를 정의하고, 음의 값들이 다른 세트를 정의) (fig. S1). - 염색체 4와 5의 경우에,
첫번째 PC는 그 두 염색체 완들(arms)에 해당했지만,
두번째 PC는 격자 무늬 패턴에 해당했다. - 그 PC 벡터의 엔트리들은 그 격자무늬 히트맵들 내 관찰된
구획에서 구획으로의 선명한 이동(sharp transitions)을 반영했다. - 더욱이, 각 염색체 내의 격자무늬 패턴들은 염색체들에 걸쳐 일관적이었다:
동일한 라벨을 보유하는 서로 다른 염색체들에서의 세트들은 상관적인 접촉 프로파일들(correlated contact profiles)을 가지도록, 그리고 다른 라벨들을 가지는 것들은 역상관적인 접촉 프로파일들(anticorrelated contact profiles)을 가지도록 라벨들(A와 B)이 부여될 수 있었다 (Fig. 3D). - 이 결과들이 시사하는 바는, 전체 게놈은
구획들에 걸쳐서 보다는
각 구획들 내에서
더 큰 상호작용이 발생하도록 2개의 공간적 구획들 (two spatial compartments)로 분할될 수 있다는 것이다. - Hi-C 데이터는
영역들(regions)이 같은 구획에 속하면 (A versus B) 그렇지 않은 경우보다
공간에서 더 가까운 경향이 있다는 것을 의미한다. - 우리는 두 구획들 간에서 번갈아 나오는(L1 and L3 in compartment A; L2 and L4 in compartment B) 염색체 14에서의 4개 유전자좌들 (L1, L2, L3, and L4)을 조사하는 3D-FISH를 사용하여 이를 테스트했다 (Fig. 3, E and F).
- 3D-FISH는, 선형의 게놈 시퀀스에서 L2가 L1과 L3 사이에 위치하는 사실에도 불구하고,
L3가 L2보다는 L1에 더 가까운 경향이 있음을 보여주었다 (Fig. 3E). - 마찬가지로, 우리는 L2는 L3보다는 L4에 더 가깝다는 것을 발견했다 (Fig. 3F).
- 22번 염색체의 연속된 4개 위치에 대해 비슷한 결과가 얻어졌다 (fig. S2, A and B).
- 보다 일반적으로,
FISH로 측정한 Hi-C reads의 수 mij와
유전자좌 i와 유전자좌 j 사이의 3D 거리 사이에 강력한 상관관계가 관찰되었고 [Spearman’s ρ = –0.916, P = 0.00003 (fig. S3)],
이는 Hi-C read count가 distance의 대용물로 사용될 수 있음을 시사한다. - Hi-C 데이터를 자세히 조사한 결과,
구획 B의 유전자좌 쌍이 주어진 게놈 거리에서 구획 A의 유전자좌 쌍보다 일관되게 더 높은 상호작용 빈도를 보였다는 것을 확인했다 (fig S4).
이는 구획 B가 더 밀집되어 있음을 시사한다 [15]. - FISH 데이터는 이 관찰과 일치한다;
구획 B의 유전자좌는 가까운 공간적 국소화 (close spatial localization)에 대한 더 강한 경향을 보였다. - 이 2개의 공간적 구획들(two spatial compartments)이
게놈의 알려진 특징들과 상응하는지 여부를 탐구하기 위해,
우리는 우리의 1-Mb correlations maps에서 식별된 구획들을 알려진 유전적 및 후성유전적 특징들과 비교하였다. - Compartment A는
presence of genes (Spearman’s ρ = 0.431, P < 10–137),
higher expression [via genome-wide mRNA expression, Spearman’s ρ = 0.476, P < 10–145 (fig. S5)],
그리고
accessible chromatin [as measured by deoxyribonuclease I (DNAseI) sensitivity, Spearman’s ρ = 0.651, P negligible]과 강하게 상관된다 [16, 17]. - Compartment A는 또한
activating chromatin marks (H3K36 trimethylation, Spearman’s ρ = 0.601, P < 10–296)와
repressive chromatin marks (H3K27 trimethylation, Spearman’s ρ = 0.282, P < 10–56)
모두의 경우에 있어서 enrichment를 보여준다 [18]. - 우리는 상기의 분석을 100 kb 해상도에서 반복한 바,
비록 compartment A와 모든 다른 게놈 및 유성유전적 특징들과의 상관관계가
강하게 유지되었지만 (Spearman’s ρ > 0.4, P negligible),
sole repressive mark인 H3K27 trimethylation과의 상관관계는 극적으로 약화되었다
(Spearman’s ρ = 0.046, P < 10–15). - 이러한 결과를 바탕으로 우리는 구획 A가
개방적이고 접근이 가능하며 활발하게 전사되는 크로마틴(open, accessible, actively transcribed chromatin)과 더 밀접하게 연관되어 있다는 결론을 내렸다. - 우리는 이상 핵형을 갖는 적백혈병 세포주인 K562 세포를 사용하여 실험을 반복했다 [19].
- 우리는 다시 두 개의 구획을 관찰했다; 이 구획들은
GM06990 세포에서 관찰된 것과 구성이 유사했고 [Pearson’s ρ = 0.732, P negligible (fig. S6)],
DNAseI sensitivity에 의해 나타난대로
개방 및 폐쇄 크로마틴 상태와 강한 상관 관계를 보였다 (Spearman’s ρ = 0.455, P < 10–154). - K562와 GM06990에서의 구획 패턴은 비슷하지만,
하나의 세포 타입에서는 개방 구획에 그리고 다른 타입들에서는 폐쇄 구획에 많은 유전자좌들이 있다 (Fig. 3H). - K562(19)의 핵형적으로 정상인 염색체에서 이러한 부조화스러운 유전자좌를 조사한 결과,
세포 타입에서의 구획 패턴과 그와 동일한 세포 타입의 크로마틴 접근성(chromatin accessibility) 간에 강력한 상관 관계가 있음을 관찰했다 (GM06990, Spearman’s ρ = 0.384, P= 0.012; K562, Spearman’s ρ = 0.366, P = 0.017). - 따라서, 따라서 고도로 재배열된 게놈에서도
공간 구획화 (spatial compartmentalization)는 크로마틴 상태 (chromatin state)와 강하게 상관관계를 갖는다.
- 우리의 결과는 게놈 전체에 걸쳐
개방 및 폐쇄 크로마틴 영역들(open and closed chromatin domains)이
핵에서 서로 다른 공간 구획을 차지한다는 것을 보여준다. - 이러한 발견은
멀리 떨어져 위치한 활성 유전자들 사이에서, 그리고
멀리 떨어져 위치한 비활성 유전자들 사이에서 모두,
그런 상호작용들의 특정한 경우들을 관찰한 개별 유전자좌에 관한 연구들을 확장한다 [8, 20–24]. - 마지막으로 우리는 구획들 내의 크로마틴 구조(chromatin structure within compartments)를 탐구하고자 했다.
- 게놈 거리 (genomic distance)의 함수로서
염색체 내 접촉 확률(intrachromosomal contact probability)의 평균 행태를 면밀히 조사하여,
게놈 전체 분포 I(s)를 계산했다. - 로그-로그 축들에 표시하면,
I(s)는 ~500kb와 ~7Mb 사이에서 눈에 띄는 冪法則 스케일링(power law scaling)을 보이며,
여기서 접촉 확률(contact probability)은 s–1 로 조정된다 (Fig 4A). - 이 범위는 알려진 크기의 개방 및 폐쇄 크로마틴 도메인에 해당한다.

Fig. 4. The local packing of chromatin is consistent with the behavior of a fractal globule.
(A) 게놈 전체에 걸쳐 평균화된 게놈 거리의 함수로서의 Contact probability (청색)는 500kb와 7Mb사이에서 (음영 영역) -1.08의 기울기로 멱법칙을 보여준다 (청록색으로 표시된 fit).
(B) equilibrium (적색) 및 fractal globule (청색)에 대한 거리의 함수로서의 접촉 확률에 대한 시뮬레이션 결과 (1개의 단량체 ~ 6개의 뉴클레오솜 ~ 1200개의 염기쌍) [10]. 프랙탈 구형체의 경우 기울기는 -1에 매우 가까워서(청록색) 우리의 예측을 확인한다 [10]. equilibrium globule의 경우 기울기는 -3/2로 이전의 이론적 기대치와 일치한다. 프랙탈 구형체의 기울기는 우리가 게놈에서 관찰한 기울기와 매우 유사하다.
(C) (상) 펼쳐진 폴리머 사슬, 4000개의 모노머(4.8Mb) 길이. 색상은 한 끝점으로부터의 거리에 해당하며, 파란색에서 청록색, 녹색, 노란색, 주황색, 빨간색까지 다양하다. (중) 평형 구형체 (equilibrium globule). 구조는 매우 얽혀 있다. 윤곽선을 따라 근처에 있는 유전자좌(유사한 색상)는 3D에서 근처에 있을 필요가 없다. (하) 프랙탈 구형체 (fractal globule). 윤곽선을 따라 근처에 있는 유전자좌는 3D에서 근처에 있는 경향이 있어 표면과 횡단면 모두에서 단색 블록이 생긴다. 구조에는 매듭이 없다.
(D) 세 가지 척도의 게놈 구조. (상) open and closed chromatin에 해당하는 두 개의 구획이 게놈을 공간적으로 분할한다. 염색체(파란색, 청록색, 녹색)는 서로 다른 영역을 차지한다. (중) 개별 염색체들이 개방형 및 폐쇄형 크로마틴 구획 사이를 앞뒤로 엮는다. (하) 싱글 메가베이스의 규모에서 염색체는 일련의 프랙탈 구형체로 구성된다.
- 冪法則 의존성(Power-law dependencies)은 폴리머와 같은 행태로부터 발생할 수 있다 [25].
- 여러 저자들이 염색체 영역(chromosomal regions)을 “평형 구형체(equilibrium globule)”로 모델화할 수 있다고 제안한 바 있다: 이는 원래 평형(equilibrium) 상태에서 빈약한 용매에 있는 폴리머를 설명하는데 사용되는 콤팩트하고 빽빽하게 매듭이 진 구성이다 [26, 27]. [역사적으로 이 특정 모델은 종종 단순히 "구체(globule)"로 언급되었다.
- 일부 저자는 “equilibrium globule”라는 용어를 사용하여 다른 구형 상태와 구별했다 (아래 참조).]
- Grosberg et al.은 계면(interphase) DNA를 포함한 폴리머가 “fractal globule”라고 설명한 수명이 긴 비평형 형태(long-lived, non-equilibrium conformation)로 자체 조직화될 수 있다는 이론을 제시하는 대체 모델을 제안했다 [28, 29]. 이 매우 콤팩트한 상태는 얽히지 않은 폴리머가 “끈에 묶인 구슬 (beads-on-a-string)” 구성의 일련의 작은 구형체들로 구겨질 때 형성된다. 이러한 비드들은 단일 구형의 구형의 구형만 남을 때까지 이후의 자발적인 구겨짐 라운드(spontaneous crumpling)에서 단량체 (monomers) 역할을 한다.
- 결과 구조는 페아노 곡선(Peano curve)과 유사하다.
페아노 곡선은 자체를 교차하지 않고 3D 공간을 조밀하게 채우는 연속적인
프랙탈 궤적 (continuous fractal trajectory)이다 [30]. - 프랙탈 구형은 매듭(knots)이 없고 [31],
예를 들어 유전자 활성화, 유전자 억제 또는 세포 사이클 동안 펼침과 재접힘을 용이하게 하기 때문에
크로마틴 세그먼트에 매력적인 구조이다. - 프랙탈 구형에서 게놈의 인접한 영역들(contiguous regions)은 원래 영역의 길이에 해당하는 크기의 공간 섹터를 형성하는 경향이 있다 (Fig. 4C).
- 반면 equilibrium globule은 매듭이 매우 많고 그러한 섹터가 없다.
- 대신 선형 및 공간 포지션들은 최대 몇 메가베이스 이후에 크게 상관 관계가 없다(Fig. 4C).
- fractal globule은 이전에 관찰된 적이 없다 [29, 31].
- Equilibrium globule 모델과 fractal globule 모델은
접촉 확률을 게놈 거리들(genomic distances)로 스케일링하는 것에 관하여 매우 다른 예측을 한다. - Equilibrium globule model은
접촉확률이 s–3/2 로 스케일할 것이며, 우리는 이를 우리의 데이터에서 관찰하지 않았다. - 우리는 분석적으로 접촉확률을 fractal globule의 경우로 도출했고,
그것이 s–1 으로 퇴조한다는 것을 발견했다; 이는 우리가 관찰한 현저한 스케일링 (s–1.08)과 밀접하게 일치한다 - Equilibrium globule 모델과 fractal globule 모델은 또한
유전자좌 쌍들 간의 3D 거리에 관하여 다른 예측을 한다
(s1/2 for an equilibrium globule, s1/3 for a fractal globule). - 비록 3D distance가 Hi-C에 의해 직접적으로 측정되지 않지만,
우리는 최근의 한 논문이 3D-FISH를 사용하여 500 kb와 2 Mb 간의 게놈 거리들의 s1/3 scaling을 보고했다는 것을 주목한다 [27]. - 우리는 fractal globules 및 equilibrium globules의 앙상블을 구성하기 위해
Monte Carlo simulations을 사용했다 (500 each). - 앙상블의 특성은
- 접촉 확률 (fractal globules의 경우 s–1, 그리고 equilibrium globules의 경우 s–3/2)과
3D 거리(fractal globules의 경우 s1/3, equilibrium globules의 경우 s1/2)에 대한
이론적으로 도출된 스케일링과 일치했다. - 이러한 시뮬레이션은 또한
얽힘의 부족 (lack of entanglements) [매듭-이론적 알렉산더 다항식(10,32)을 사용하여 측정]과 프랙탈 구형체 내의 공간 섹터 형성을 보여주었다 (Fig. 4B). - 우리는 수 메가베이스의 규모에서
데이터가 크로마틴 조직에 대한 fractal globule model과 일치한다는 결론을 내렸다. - 물론, 다른 형태의 규칙적인 조직이 유사한 결과를 가져올 가능성을 배제할 수는 없다.
- 여기서는 비교적 대규모의 상호작용에 초점을 맞추었다.
- Hi-C는 또한 number of reads를 늘려 더 미세한 규모에서 포괄적인 게놈-전체의 상호작용 맵을 구성하는 데 사용할 수 있다.
- 이를 통해 enhancers, silencers 및 insulators 간의 특정 장거리 상호작용 (long-range interactions)을
매핑할 수 있다 [33–35]. - 분해능을 n배 높이려면 number of reads를 n2배로 늘려야 한다.
- 시퀀싱 비용이 낮아짐에 따라 더 미세한 상호작용을 감지하는 것이 점점 더 가능해질 것이다.
- 또한 크로마틴 면역침전(chromatin immunoprecipitation)이나 하이브리드 캡처(hybrid capture)를 사용하여
게놈 서브 세트들에 집중할 수도 있다 [36, 37].


'Coffee Genetics' 카테고리의 다른 글
| 커피의 다양성과 게놈 진화 (10) | 2024.08.11 |
|---|---|
| 아라비카 커피의 염색체-규모 어셈블리 - 染色體 異常 및 交換 (0) | 2024.07.24 |
| Oxford Nanopore Technologies Sequencing - 3세대 시퀀싱 (1) | 2024.07.21 |
| Complete Genomics cPAL Sequencing - 3세대 시퀀싱 (0) | 2024.07.19 |
| PacBio SMRT Sequencing - 3세대 DNA 시퀀싱 (0) | 2024.07.17 |




댓글